NWChem
[1]:
%matplotlib inline
import os, bz2
import exatomic
from exatomic import nwchem
[4]:
out = nwchem.Output(exatomic.base.resource("nw-ch3nh2-augccpvdz.out"))
out.head()
0: argument 1 = ch3nh2.nw
1:
2:
3:
4: ============================== echo of input deck ==============================
5: echo
6: start ch3nh2
7: title ch3nh2
8:
9: geometry units angstrom noautoz nocenter
[5]:
out.tail()
2954: K. Hirao, R. A. Kendall, J. A. Nichols, K. Tsemekhman, K. Wolinski,
2955: J. Anchell, D. E. Bernholdt, P. Borowski, T. Clark, D. Clerc, H. Dachsel,
2956: M. J. O. Deegan, K. Dyall, D. Elwood, E. Glendening, M. Gutowski, A. C. Hess,
2957: J. Jaffe, B. G. Johnson, J. Ju, R. Kobayashi, R. Kutteh, Z. Lin,
2958: R. Littlefield, X. Long, B. Meng, T. Nakajima, S. Niu, L. Pollack, M. Rosing,
2959: K. Glaesemann, G. Sandrone, M. Stave, H. Taylor, G. Thomas, J. H. van Lenthe,
2960: A. T. Wong, Z. Zhang.
2961:
2962: Total times cpu: 3.9s wall: 4.1s
[6]:
out.parse_atom()
out.atom
[6]:
| tag | Z | x | y | z | symbol | Zeff | frame | |
|---|---|---|---|---|---|---|---|---|
| atom | ||||||||
| 0 | C | 6 | 1.409566 | 0.020987 | 0.000000 | C | 6 | 0 |
| 1 | N | 7 | -1.363889 | -0.135772 | 0.000000 | N | 7 | 0 |
| 2 | H | 1 | 1.969196 | 2.003298 | 0.000000 | H | 1 | 0 |
| 3 | H | 1 | 2.134053 | -0.913404 | 1.686645 | H | 1 | 0 |
| 4 | H | 1 | 2.134053 | -0.913404 | -1.686645 | H | 1 | 0 |
| 5 | H | 1 | -2.035202 | 0.730039 | -1.562856 | H | 1 | 0 |
| 6 | H | 1 | -2.035202 | 0.730039 | 1.562856 | H | 1 | 0 |
[7]:
out.parse_basis_set()
out.basis_set.tail()
[7]:
| shell | L | alpha | d | set | frame | |
|---|---|---|---|---|---|---|
| function | ||||||
| 52 | 0 | 0 | 0.44460 | 0.478148 | 2 | 0 |
| 53 | 1 | 0 | 0.12200 | 1.000000 | 2 | 0 |
| 54 | 2 | 0 | 0.02974 | 1.000000 | 2 | 0 |
| 55 | 3 | 1 | 0.72700 | 1.000000 | 2 | 0 |
| 56 | 4 | 1 | 0.14100 | 1.000000 | 2 | 0 |
[8]:
out.parse_basis_set_order()
out.basis_set_order.tail()
[8]:
| center | shell | L | ml | frame | |
|---|---|---|---|---|---|
| chi | |||||
| 86 | 6 | 3 | 1 | -1 | 0 |
| 87 | 6 | 3 | 1 | 0 | 0 |
| 88 | 6 | 4 | 1 | 1 | 0 |
| 89 | 6 | 4 | 1 | -1 | 0 |
| 90 | 6 | 4 | 1 | 0 | 0 |
[9]:
out.parse_frame()
out.frame
[9]:
| atom_count | total_energy | |
|---|---|---|
| frame | ||
| 0 | 7 | -95.229252 |
[10]:
out.parse_momatrix()
out.momatrix.shape
[10]:
(8281, 4)
[11]:
out.parse_orbital()
out.orbital.tail()
[11]:
| energy | frame | occupation | vector | x | y | z | spin | group | |
|---|---|---|---|---|---|---|---|---|---|
| orbital | |||||||||
| 86 | 3.034495 | 0 | 0.0 | 86 | 0.079 | -3.900000e-14 | -3.900000e-14 | 0 | 0 |
| 87 | 3.059546 | 0 | 0.0 | 87 | -0.370 | 3.000000e-13 | 3.000000e-13 | 0 | 0 |
| 88 | 3.074835 | 0 | 0.0 | 88 | 0.095 | -6.900000e-13 | -6.900000e-13 | 0 | 0 |
| 89 | 3.380757 | 0 | 0.0 | 89 | -0.800 | 1.800000e-13 | 1.800000e-13 | 0 | 0 |
| 90 | 3.634046 | 0 | 0.0 | 90 | -0.670 | -2.200000e-13 | -2.200000e-13 | 0 | 0 |
[12]:
uni = out.to_universe()
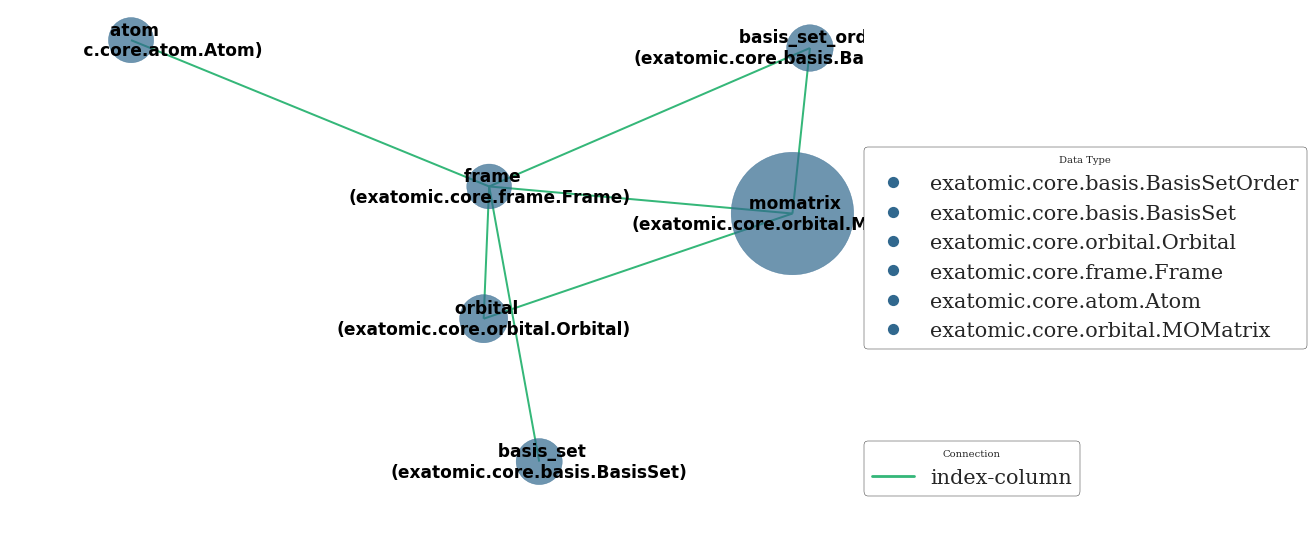
[13]:
uni.network()
[13]:
<networkx.classes.graph.Graph at 0x7f82eb582630>
[14]:
uni.compute_atom_two()
uni.atom_two.bonded
[14]:
| atom0 | atom1 | dr | bond | |
|---|---|---|---|---|
| two | ||||
| 0 | 0 | 1 | 2.777882 | True |
| 1 | 0 | 2 | 2.059791 | True |
| 2 | 0 | 3 | 2.059791 | True |
| 3 | 0 | 4 | 2.059791 | True |
| 9 | 1 | 5 | 1.908614 | True |
| 10 | 1 | 6 | 1.908614 | True |
[15]:
uni.orbital[uni.orbital['occupation'] != 0.0]
[15]:
| energy | frame | occupation | vector | x | y | z | spin | group | |
|---|---|---|---|---|---|---|---|---|---|
| orbital | |||||||||
| 0 | -15.544670 | 0 | 2.0 | 0 | -0.72 | -3.700000e-15 | -3.700000e-15 | 0 | 0 |
| 1 | -11.248320 | 0 | 2.0 | 1 | 0.75 | 1.300000e-15 | 1.300000e-15 | 0 | 0 |
| 2 | -1.170266 | 0 | 2.0 | 2 | -0.45 | 1.200000e-13 | 1.200000e-13 | 0 | 0 |
| 3 | -0.893624 | 0 | 2.0 | 3 | 0.46 | 1.200000e-13 | 1.200000e-13 | 0 | 0 |
| 4 | -0.668444 | 0 | 2.0 | 4 | -0.43 | -3.800000e-13 | -3.800000e-13 | 0 | 0 |
| 5 | -0.597788 | 0 | 2.0 | 5 | 0.40 | 1.100000e-13 | 1.100000e-13 | 0 | 0 |
| 6 | -0.555492 | 0 | 2.0 | 6 | 0.18 | 9.200000e-13 | 9.200000e-13 | 0 | 0 |
| 7 | -0.522872 | 0 | 2.0 | 7 | 0.51 | -1.800000e-12 | -1.800000e-12 | 0 | 0 |
| 8 | -0.385592 | 0 | 2.0 | 8 | -0.45 | -1.600000e-13 | -1.600000e-13 | 0 | 0 |
[16]:
uni.add_molecular_orbitals(field_params={'rmin': -20, 'rmax': 20, 'nr': 51})
Warning: Check spherical shell parameter for nwchem molecular orbital generation
Evaluating 91 basis functions once.
Timing: compute orbitals - 3.56s.
[17]:
exatomic.UniverseWidget(uni)
[ ]:
[ ]:
[ ]: